
依数性
溶质的溶解是个物理过程。溶解的结果是溶质和溶剂的某些性质发生了变化,这些性质变化(即溶液性质)可归纳为两类:一类是由溶质的本性决定的,如密度、颜色、导电性、酸硷性等;而另一类性质是由溶质粒子数目的多少决定的,如溶液的蒸汽压下降、沸点升高、凝固点下降和渗透压等,尤其在非电解质稀溶液中的这些性质与溶质的本性无关,而只与溶质数量有关,故称为溶液的“依数性”。
基本介绍
- 中文名:依数性
- 外文名:colligative properties
- 别称:依数性质
- 提出者:拉乌尔,范特霍夫,亨利
- 提出时间:1887
- 套用学科:化学
- 适用领域範围:溶液的性质
- 适用领域範围:测定含量
发现简史
拉乌尔(又译娄尔特Raouh)和普费弗(Pfeffer)的基本研究工作奠定了近代溶液理论的基础。溶液的依数性的发现是伴随着溶液的渗透压、凝固点下降、沸点升高和蒸汽压下降等现象的发现和理论的提出而逐渐发展的。
渗透压的发现
18 世纪中叶,安托万·诺莱(Abbé Nollet) 发现:水能自然地透过猪膀胧扩散到酒精溶液内。他首次揭示了膜分离现象,但限于当时的认识能力,尚不知此为渗透压问题。首先探明渗透压力问题的是特劳伯(M.Traube),他在1867年实验人造植物细胞时,发现了一种由亚铁氰化铜(cupric ferrocyanide)製成的半透膜。但由于这种半透膜不够坚固的原因,欲测定渗透压的绝对数值颇不容易。1877年莱比锡大学植物教授威廉·普费弗将亚铁氰化铜沉积在素瓦罐壁有孔盆上,製得适用而强固的半透膜,它可以只让水通过而不让溶解的糖透过去,于是普费弗用这种半透膜测量了多种溶液的渗透压,由此发现渗透压与溶液浓度成正比。但是除植物学家外,这个结论并未众人所熟知。1884年阿姆斯特丹植物学家德·福利斯(de Vries)特别研究了等渗溶液,并将其研究告诉范特霍夫后,范特霍夫立即看出它在物理化学上的伟大意义。范特霍夫结合普费弗的实验数据和查尔斯等定律并运用热力学证明了:溶质在溶液中产生的渗透压等于占有和溶液相同体积的气体产生的压力。其渗透压定律便可以用理想气体状态方程推算。但在譬如盐酸、强硷和盐等溶液中好像有的代表二倍分子数,当时,范特霍夫发表其溶液学说时,尚不明白为什幺有此类现象,于是用一个因子“i”放入方程式中,以表示反常大的渗透压。
凝固点下降的发现
盐水比淡水的凝固点低些的事实,早在1788年卡文迪许(又译凯文第旭)的助手布莱格登(Blagden)就首先研究过,他知道同一化合物的溶液的凝固点下降值和浓度有比例关係,但此后100年,此问题尚未有合理的解释。直至1878年拉乌尔发表了关于溶液凝固点的第一篇着作,1881年乌拉尔教授主要用有机物进行多次实验,并把结果在1882年列成一张表,表中所示100克水中含有1克的物质的溶液,其凝固点的下降与此物质的相对分子质量的乘积是一个常数。他说:“这似乎证明,在大多数情况下,有机化合物的分子,被溶解作用简单的分开,成为相同的状态,对水的物理性质产生同样的影响。”
他把凝固点下降数值称为“下降係数”(depression coefficient),把下降係数和分子量的乘积的常数,称为“分子下降”(molecular depression),经其种种实验,他发现有机化合物的分子下降乃是分子其中各个原子的“原子下降”的平均数。至于原子下降是原子的本性,可算出C、H、O、N的原子下降是15、15、30、30,一常规有机化合物CpHqOrNs的分子下降,为(15p+15q+30r+30s)/(p+q+r+s),他说只要测出下降係数,便可推算出一个化合物的分子式。举草酸为例,通过燃烧和分析等,草酸的实验式CHO2,其可能分子式C2H2O4、C3H3O6等,但无论用那个分子式,算出的分子下降都是22.5。乌拉尔测得草酸下降係数为0.255。故其大概分子量22.5/0.255=88.3,故知C2H2O4(分子量为90)为草酸的正当分子式,这种发现立即引起有机化学家的注意,因为以前无可靠方法测定不挥发物质的分子量,可以在溶液中测定了。但是,拉乌尔寻求某些化合物的分子下降值时,有时竟然有两个,且一个差不多是另一个的两倍,发现醋酸的值有18和39,甲酸有14和28,硝基苯有36和72,二溴化乙烯有58和118等等,按照乌拉尔实验,许多盐类、强酸、和强盐基的为何都有异常大的凝固点下降和沸点上升,是后来从其他方面才得到的解释。
沸点升高的发现
沸点升高的现象也久为人知,1822年法拉第(Faraday)、1824年格里菲斯(Griffiths)和1835年勒格朗(Legrand)也都考察过,尚不得要领。及拉乌尔做过凝固点下降和其他实验后,才证明沸点上升的规律和凝固点下降相似。
蒸气压下降的发现
早在1822年就已经知道溶液的蒸气压,比纯溶剂的低,但到1855年Wüllner才用实验发现,如果溶质是不挥发的,则溶液的蒸气压下降与溶解物质的数量有比例关係。1886-1887年拉乌尔证明:“1克分子固定的、非言物质,溶解在100克分子任何挥发性液体中,液体蒸气压的降低是其蒸气压数字的接近不变的分数,这分数大约是0.0105。”这又是一个用溶液来求相对分子质量的方法。1887年拉乌尔用萜烯、硝基苯、苯胺、甲酯和苯甲酸乙酯5种化合物,取每种各异数量,放于水、二硫化碳、四氯化碳、丙酮、戊烯、苯等11种溶剂中。其结果都是:只要在100溶剂分子中的溶质分子数不大于15,溶液蒸气压下降数值与溶质的分子数目有比例关係。又以分子重量为比例,将各物质溶于等容的同一溶剂中,蒸气压下降的数值都相等。又发现一个规则:蒸气压的下降值与原来蒸气压的比值即下降率等于溶质分子数与溶液分子总数的比值。由此与1887年发现拉乌尔定律,并于理论方面找出凝固点下降和沸点上升与蒸气压下降的关係,此时范特霍夫的溶液学说也已经成立,又找出它们三者和渗透压的关係,于是在溶液中测定有机分子量有了确切的理论基础。
电解质理论的发现
对于拉乌尔曾经注意到的异常的分子下降和范特霍夫对盐类溶液的异常偏大的渗透压,首次完全给出解释的是阿侖尼乌斯(Arrhenius)的电离理论。但这个学说套用在弱电解质上最为相宜,不能套用于强电解质,虽然有试验的公式来表示强电解质的行为,但都无理论上的依据。直到1923德国化学家德拜(Debye)和休克尔(Hückel)提出的离子互吸理论才将其合理化。但较浓的溶液中离子相隔较近,所引起的完全反乎电离的行为不断增大,至今电解质溶液理论仍然在不断完善中。
相关概念
稀溶液与纯溶剂相比某些物理性质会有所变化如蒸气压下降、凝固点降低、沸点升高和渗透压的产生。这是多组分系统中化学势随组分数而表现出来的自身变化规律,溶液的依数性只有在溶液的浓度很稀的时候才有规律,而且溶液越稀,其依数性的规律性越强。
除此之外,依数性还受到溶质的电解性和挥发性的影响。
溶液按电解质在溶液中的电离程度可将溶液分为非电解质溶液和电解质溶液,其中电解质溶液又有强电解质溶液和弱电解质溶液之分;在一定温度下,不同物质有不同的蒸气压。通常把常温下蒸汽压较低的物质称为难挥发物质,如甘油、葡萄糖、食盐等;蒸气压较高的物质称为易挥发物质,如苯、乙醇、碘等。所以蒸气压越大,越易挥发,沸点越低。
由于至今电解质理论和浓溶液理论论仍在不断完善中,而挥发性溶质的依数性偏差波动很大,故本词条着重讨论溶剂为难挥发非电解质的稀溶液的依数性。
蒸气压下降
在一定温度下,将纯液体置于真空容器中,当蒸发速度与凝聚速度相等时,液体上方的蒸气所具有的压力称为该温度下液体的饱和蒸气压,简称蒸气压。任何纯液体在一定温度下都有确定的蒸气压,且随温度的升高而增大。下面有两个实验可直观地说明蒸气压下降的具体现象。
实验验证
实验一,见右图:在一封闭容器,放一杯纯水和浓溶液(浓糖水)。经过一段时间后,发现糖水的液面面上升了,纯水的液面下降。水蒸气会不断的凝聚于糖水中,直到水杯中的水全部转移到糖水杯中为止。这个实验被称为溶剂转移实验。
这只有假设糖水的蒸气压比水的蒸气压低才显得合理,因为在这个密闭的容器里,有三个水的蒸气压:纯水的蒸气压代表纯水的蒸发速度,密闭容器的空气中的水蒸气压代表水的凝聚速度,以及糖水的蒸气压代表糖水的蒸发速度。显然纯水的蒸发速度大于水的凝聚速度,导致纯水的液面下降;糖水的蒸发速度小于水的凝聚速度,导致液面的上升;这说明了纯水的蒸气压大于密闭容器中的水蒸气压大于的糖水的蒸气压。而且这一实验通过水的转移时间,可以证明溶液浓度越大,溶液的蒸气压下降的越显着。
实验二,初始状态见左下图的装置中,左管是纯水,右管为葡糖糖溶液,两管由U形压强计相连,U型管中的水银高度相同。关闭阀门,置水浴恆温。然后打开阀门,一定时间后,见右下图,可观察到压强计的水银面右柱高于左柱,表明葡萄糖溶液的蒸气压小于水的蒸气压。而且这一实验通过水银液差,可以证明溶液浓度越大,溶液的蒸气压下降的越显着。


现象解释
当纯溶剂溶解一定量难挥发溶质(如蔗糖溶于纯水时,萘溶于苯中)时,在同一温度下,溶液的蒸汽压总是低于纯溶剂的蒸汽压。这种现象称为蒸汽压下降,即
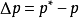
式中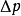 ---溶液的蒸气压下降值;
---溶液的蒸气压下降值;
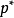
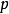
稀溶液蒸气压下降的原因可以从以下两个方面来解释,如下图所示:一方面,溶质分子占据着一部分溶剂分子的表面,在单位时间内逸出页面的溶剂分子数目相对减少;另一方面,在溶剂中加入了难挥发的非电解质后,每个溶质分子与若干个溶剂分子相结合,形成了溶剂化分子,溶剂化分子一方面束缚了一些能量较高的溶剂分子。因此,达到平衡时,溶液的蒸气压必定低于纯溶液的蒸气压,且浓度越大,蒸气压下降越多。这也是解释沸点升高和凝固点下降更为根本的原因。

数学推导
对于两组分溶液,由道尔顿分压定律,注意在体系的气相中纯溶剂蒸气压代表了总的压强,而溶液的蒸气压由于非电解质分子的阻隔和溶剂化作用,相当于总气压的分压: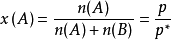
对上式进行变换, ,因为摩尔分数总是小于等于1的,所以溶液的蒸气压必定低于纯溶剂的蒸气压。而且公式 也称为拉乌尔定律,其证明了蒸气压下降与溶液组成的不同量度之间的关係。
,因为摩尔分数总是小于等于1的,所以溶液的蒸气压必定低于纯溶剂的蒸气压。而且公式 也称为拉乌尔定律,其证明了蒸气压下降与溶液组成的不同量度之间的关係。
拉乌尔定律
1887年,法国物理学家拉乌尔(F.M.Raoult)研究了溶质对纯溶剂蒸气压的影响,根据大量实验结果提出下列三个观点,这些规律称为拉乌尔定律,拉乌尔定律证明了蒸气压下降与溶液组成的不同量度之间的关係,在此一一证明论述。
在一定温度下,难挥发非电解质稀溶液的蒸气压,等于纯溶剂的蒸气压乘以溶剂A在溶液中的摩尔分数(一说物质的量分数)。其数学表达式: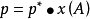
用x(B)表示难挥发非电解质的物质的量分数,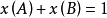
所以,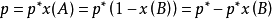
用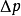 表示溶液的蒸气压下降值,
表示溶液的蒸气压下降值,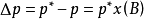
上式表明,拉乌尔定律又可以表示为:在一定温度下,难挥发非电解质稀溶液的蒸气压下降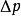 与溶质B摩尔分数成正比。
与溶质B摩尔分数成正比。
其数学表达式为: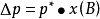
对于两组分溶液: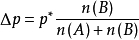
当溶液很稀时,溶质B的物质的量小到可以忽略不计:
即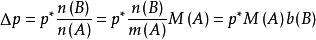
当温度一定时,p*和M(A)都是常数,其乘积用K表示,则: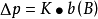
因此,拉乌尔定律又可以表述为,在一定温度下,难挥发非电解质稀溶液的蒸气压下降,近似地与溶质B的质量摩尔浓度成正比,而与溶质的本性无关。
套用领域
溶液凝固点下降在冶金工业中具有指导意义。一般金属的Kf都较大,例如Pb的Kf≈130 K kg/mol,说明熔融的Pb中加入少量其它金属,Pb的凝固点会大大下降,利用这种原理可以製备许多低熔点合金。金属热处理要求较高的温度,但又要避免金属工件受空气的氧化或脱碳,往往採用盐熔剂来加热金属工件。例如在BaCl2(熔点1236 K)中加入5%的NaCl(熔点1074 K)作盐熔剂,其熔盐的凝固点下降为1123 K;若在BaCl2中加入22.5%的NaCl,熔盐的凝固点可降至903 K。
沸点升高
现象内容
当液体的蒸气压等于外压时,液体的汽化将在其表面和内部同时发生,这种汽化过程称为液体的沸腾,此时再给体系加热,只会使更多的液体汽化,而体系的温度不会上升。沸点Tb(boiling point)是当纯液体或溶液的蒸气压与外界大气压相等时,溶液沸腾的温度。值得注意的是,沸点与外压有关,外压越大,沸点越高。
沸点升高的现象特徵可以从右图的纯水与水溶液的相图观察得出。图中,纵坐标为蒸气压,横坐标为体系温度。ab曲线为纯水的蒸气压随温度变化曲线,a‘b’曲线为水溶液的蒸气压随温度变化的曲线。从a‘b’曲线在ab曲线下方,可以看出水溶液的蒸气压在任何温度都小于纯水的蒸气压。
如果未指明外界压力,默认外界大气压为101.325 kPa。在373.15K时,水的蒸气压等于外界大气压,所以水的沸点是373.15K(100℃)。在右图中,当纵坐标等于外界大气压(101.3kPa)时,水的正常沸点Tb*小于水溶液的沸点Tb。这种溶液的沸点高于溶剂沸点的现象为溶液的沸点升高。
溶液的沸点升高的数值ΔTb等于溶液的沸点Tb与纯溶剂的沸点Tb*之差:
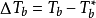
现象解释
之所以会出现溶液的沸点高于纯溶剂沸点的现象,是因为在溶剂加入少量难挥发的非电解质后,所形成的溶液由于蒸气压下降,其蒸气压低于外界大气压,故溶液达到纯溶剂的沸点时,仍不能沸腾。于是要使溶液沸腾,必须继续升高温度,使得溶液的蒸气压达到外界压力,其温度已经超过纯溶剂的沸点,所以这类溶液的沸点总是比纯溶剂的沸点高。
由于溶液的沸点升高的根本原因是溶液的蒸气压下降,所以,溶液浓度越大,则蒸气压下降的越多,于是沸点升高得越多。而且溶液沸点升高是由于溶液蒸气压下降引起的,对于难挥发非电解质的稀溶液,既然蒸气压下降和溶液的质量摩尔浓度b(B)成正比,这类溶液的沸点升高也应和质量摩尔浓度有联繫。类似地,拉乌尔(Raoult)根据依数性指出:难挥发非电解质稀溶液的沸点升高也近似地与溶质B的质量摩尔浓度成正比,即
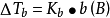
式中:Kb称为沸点升高常数(boiling point constant),单位为K·kg·mol-1或℃·kg·mol-1,这个数值只取决于溶剂,而与溶质无关。沸点升高常数的计算公式:
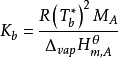
式中: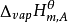 是纯溶剂的摩尔汽化热,R为摩尔气体常量,Tb*为纯溶剂的沸点,MA为溶剂A的摩尔质量。
是纯溶剂的摩尔汽化热,R为摩尔气体常量,Tb*为纯溶剂的沸点,MA为溶剂A的摩尔质量。
溶剂 | 沸点Tb/K (t/℃) | Kb/K·kg·mol-1 |
水 | 373.15(100) | 0.512 |
苯 | 353.25(80.1) | 2.53 |
乙醇 | 351.65(78.5) | 1.22 |
乙酸 | 391.05(117.9) | 3.07 |
丙酮 | 329.35(56.2) | 1.71 |
四氯化碳 | 349.70(76.6) | 5.03 |
萘 | 491.15(218.0) | 5.80 |
酚 | 454.35(181.20) | 3.60 |
乙醚 | 307.85(34.70) | 2.02 |
环己烷 | 354.15(81) | 2.79 |
樟脑 | 481.15(208) | 5.95 |
参考资料来源
数学推导
挥发性溶剂A在正常沸点Tb*时存在下述液-气平衡:A(1,Tb*,pΘ)⇌A(g,Tb*,pΘ)
根据相平衡关係,其平衡条件为μAΘ(1,Tb*)=μAΘ(g,Tb*) (1)
当向溶剂A加入非挥发性溶质B后,溶液的正常沸点为Tb。此时的液-气平衡条件为
μAΘ(1,Tb*)+RTblnxA=μAΘ(g,Tb) (2)
因为△vapGmΘ,A(Tb)=μAΘ(1,Tb*)-μAΘ(g,Tb*)
(1)、(2)两式可以整理为:△vapGmΘ,A(Tb)/Tb*=0,△vapGmΘ,A(Tb)/Tb=RTblnxA
相减得△vapGmΘA(Tb)/Tb*-△GmΘ,A(Tb)/Tb=-RTblnxA
当温度温度变化较小,吉布斯-亥姆霍兹公式的积分形式:
△G(T2)/T2-△G(T1)/T1=-△H(T2-T1)/T1T2
将此公式套用到上式,整理可得:
-lnxA=△vapHmΘ,A(Tb-Tb*)/RTbTb*≈△vapHmΘ,A· ΔTb/R(Tb*)2,其中TbTb*近似为(Tb*)2。
对于二元稀溶液,xA→1,xB→0,-lnxA=-ln(1-xB)≈xB≈nB/nA
n为溶液中A、B的物质的量,于是可写成:ΔTb=nBR(Tf*)2/nA△vapHmΘ,A
若用b(B)表示质量摩尔浓度,M表示摩尔质量,m表示质量,又可写成:
ΔTb=MAmBR(Tb*)2/MBmA△vapHmΘ,A=b(B)·MAR(Tb*)2/△vapHmΘ,A=Kb·b(B)
以上,是此公式 的证明过程,即拉乌尔根据依数性指出:难挥发非电解质稀溶液的沸点升高近似地与溶质B的质量摩尔浓度成正比的数学证明过程。
套用领域
钢铁工件进行氧化热处理就是套用沸点升高原理。用每升含550~650 g NaOH和100~150 g NaNO2的处理液,其沸点高达410~420 K。
在钢铁冶炼工业中,通过观测钢水的沸点来确定其他组分的含量在钢铁工业生产中,技术员为了配比一定比率的固溶体需要不断的取样测定,不仅重複劳动、工作量大,而且高温作业採样会有很大的潜在危险,于是技术员通过观测安装在熔炉中温度测量仪测定每一个状态时的沸点,就可以确定即时合金中的其他金属的含量,对合金生产起到关键的调控作用.这就依据依数性的沸点上升原理,在纯铁水中加入另一种金属后沸点会升高,不同的组分含量就对应相应的沸点,通过沸点的变化值就可计算出在某一沸点时另一种金属的含量,对钢铁合金的调节既方便又简捷。
沸点升高法可以测定的是聚合物的数均分子量,原理:在溶剂中加入不挥发性溶质后,溶液的蒸汽压下降,导致溶液的沸点高于纯溶剂,这些性质的改变值正比于溶液中溶质分子的数目。
计算公式为: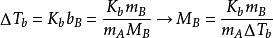
凝固点下降
凝固点是物质在一定的外压下,其液相蒸气压和固相蒸气压相等,此时液体的凝固和固体的熔化处于平衡状态,从溶液中开始析出溶剂晶体时的温度的温度。溶液的凝固点实际上就是溶液中的蒸气压与纯固体溶剂的蒸气压与纯固体溶剂的蒸气压相等时的温度。
现象内容
凝固点下降的现象特徵可以从纯水与溶液的步冷曲线观察出。物质的温度随时间而变化的曲线,叫做步冷曲线。在步冷曲线中,纵坐标为温度,横坐标为时间,如右图所示。
曲线(1)是纯水的步冷曲线,AB段是H2O的液相,温度不断下降。B点开始结冰,BC段表示温度不变;在C点处纯水全部结冰,CD段表示冻的温度不断下降。
曲线(2)是稀溶液的步冷曲线,.A’B’段是液相,温度不断下降;B’点低于273K,是溶液的冰点,B’C’段温度不恆定,有冰析出,随着溶剂不断结晶析出,溶液浓度将不断增大,凝固点也将不断下降,所以凝固点不恆定;C’点时,冰和溶质一同析出,且二者具有固定的比例。即和此时溶液的比例相同。C’点的温度称为低共熔点,从C’点析出的冰盐混合物,叫低共熔混合物,即两种或两种以上物质形成的熔点最低的混合物。
曲线(3)是同溶质的浓溶液的步冷曲线。观察三条曲线,不难看出B”的温度比B’温度低,B‘的温度比B温度低,即溶液的冰点下降,且随着溶液的浓度增加,冰点更低。这种溶液的凝固点低于纯溶剂的凝固点的现象称为溶液的凝固点下降。
除此之外,还可以从右图所示的相图观察。这时体系是由溶液(液相)溶剂(固相)和溶剂(气相)所组成。A点是纯水的凝固点Tf*(273.15K),此时水的蒸气压与冻的蒸气压相等为610.5Pa(4.58 mm Hg),若其液、固两相的蒸气压不相等,则两相不能共存。A点的左边的曲线表示冰,其曲线斜率大,意味着冻的蒸气压随温度变化大。当273.15K时,冻的蒸气压仍为610.5 Pa,而溶液的蒸气压曲线在A点下面,数值小于610.5Pa,两相的蒸气压相等,若两者接触则冰溶化。可见只有在273.15 K以下的某个温度时,溶液和冰才能共存。可见溶液的凝固点总是比纯溶剂的低,这就是凝固点下降的现象。
即有溶液的凝固点下降的数值ΔTf等于纯溶剂的沸点Tf*与溶液的沸点Tf与之差,即
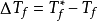
注意事项

但是有学者指出,上面的凝固点降低的图解显然错了。标準凝固点或标準熔点是指标準态压力下液态与固态处于平衡的温度 。纯水的凝固点和水溶液的凝固点应当是左图上的B点和F点。凝固点下降值则为两点之差 ,但绝不是上图的稀溶液的气 - 液曲线向左下方延至其液气 - 固曲线的交叉点 ,那个点是水的三相点而非凝固点。目前只有少数教材所用的图解是对的,因为多数教材将沸点升高和凝固点下降放到一个图中讨论了。
误差在于水的三相(O)点是指纯水在固液气三相同时存在达到平衡时其所对应的状态,温度为273.16K,压强为611.775Pa。水的凝固点(B)点是指饱和空气的水溶液在1atm下凝固时对应的状态,温度为273.15K,压强为101.325Pa。虽然二者温度相差0.01K,而压强相差极大。图中,OA、OB和OC分别表示纯溶剂水的气-液,液-固和固-气两相平衡线,依据纯组分两相平衡热力学,它们都满足克拉贝龙方程,其中OA和OB线还满足克劳修斯-克拉贝龙方程;由于水凝固时体积增大,所以OB线斜率为负值,O点为三相点;在OB线左边的虚线EF为稀溶液的凝固点随外压的变化。
现象解释

溶液的凝固点下降原因的与沸点升高一样,本质上都是由于溶液的蒸气压下降所引起的。这是因为溶质分子和溶剂化分子对冻的蒸发阻隔作用不大,即对冻的蒸气压影响不大,所以溶液的蒸气压下降的多,其蒸气压必然低于冻的蒸气压,即溶液的凝固速度小于冻的蒸发速度,这样原本的凝固点的温度不能使得固液平衡了。要使溶液凝固,就必须让溶液的蒸气压和冻的蒸气压相等,即此时的温度应该还要比原先凝固点还要低。
若进一步降低溶液的温度,溶液和冻的蒸气压会同时下降,且由于冻的蒸气压的下降率比水溶液的蒸气压大,溶液的蒸气压和冻的蒸气压会在凝固点以下的某一温度下达到平衡状态。如溶液凝固点下降图所示,此时溶液的蒸发速度与冻的蒸发速度与溶液的凝聚速度相等后,对应温度即溶液的现在的凝固点,这就是凝固点下降的原理。
既然溶液的凝固点下降也是由于溶液蒸气压下降引起的对于难挥发非电解质的稀溶液,那幺类似的,这类溶液的凝固点下降也应和质量摩尔浓度b(B)有联繫。拉乌尔根据依数性指出:对于难挥发非电解质的稀溶液,凝固点下降ΔTf都和溶液质量摩尔浓度成正比,即
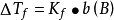
式中:Kf称为凝固点下降常数(freezing point constant),单位为K·kg·mol-1或℃·kg·mol-1,这个数值只取决于溶剂,而与溶质无关。下面是凝固点下降常数Kf的计算公式:
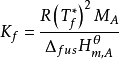
式中: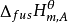 是纯溶剂的摩尔熔融焓,R为摩尔气体常量,Tb*为纯溶剂的沸点,MA为溶剂A的摩尔质量。
是纯溶剂的摩尔熔融焓,R为摩尔气体常量,Tb*为纯溶剂的沸点,MA为溶剂A的摩尔质量。
溶剂 | 沸点Tf/K (t/℃) | Kf/K·kg·mol-1 |
水 | 273.15(0) | 1.86 |
苯 | 278.50(5.35) | 5.12 |
硝基苯 | 278.85(5.70) | 6.90 |
乙酸 | 289.75(16.6) | 3.90 |
环己醇 | 279.69(6.54) | 39.30 |
四氯化碳 | 250.20(-22.95) | 29.8 |
萘 | 80.30(353.45) | 5.12 |
酚 | 313.15(40) | 7.27 |
环己烷 | 279.65(6.5) | 20.20 |
樟脑 | 351.15(78) | 40.00 |
参考资料来源
Kf和Kb的数值均不是在b(B)=1mol·kg-1时测定的,因为许多物质当其质量摩尔浓度远未到1mol·kg-1时,拉乌尔定律已不适用。此外,还有许多物质的溶解度很小,根本不能形成1mol·kg-1溶液,实际Kb和Kf值是从稀溶液的一些实验结果推算而得出的。
数学推导
纯溶剂A在正常凝固点Tf*时存在下述固-液平衡:A(s,Tf*,pΘ)⇌A(1,Tf*,pΘ)
根据相平衡关係,其平衡条件为μAΘ(s,Tf*)=μA(1,Tf*) (1)
当向溶剂A加入溶质B后,溶液的正常凝固点为Tf。设只有A结晶析出,B不结晶析出。则体系固-液平衡为
μAΘ(s,Tf*)=μAΘ(1,Tf) +RTblnxA (2)
因为△fusGmΘ,A(T)=μAΘ(1,T)-μAΘ(g,T)
(1)、(2)两式可以整理为:△fusGm,A(Tf*)/Tf*=0,△fusGm,A(Tf)/Tf=-RTflnxA
相减得△fusGm,A(Tf)/Tf-△fusGm,A(Tf*)/Tf*=-RTflnxA
当温度温度变化较小,吉布斯-亥姆霍兹公式的积分形式:
△G(T2)/T2-△G(T1)/T1=-△H(T2-T1)/T1T2
将此公式套用到上式,整理可得:
-lnxA=△fusHmΘ,A(Tf*-Tf)/RTfTf*≈△fusHmΘ,A· ΔTf/R(Tf*)2 ,其中TfTf*近似为(Tf*)2。
对于二元稀溶液,xA→1,xB→0,则-lnxA=-ln(1-xB)≈xB≈nB/nA
n为溶液中A、B的物质的量,于是可写成:ΔTf=nBR(Tf*)2/nA△fusHmΘ,A
若用b(B)表示质量摩尔浓度,M表示摩尔质量,m表示质量,又可写成:
ΔTf=MAmBR(Tf*)2/MBmA△fusHmΘ,A=b(B)·MAR(Tf*)2/△fusHmΘ,A=Kf·b(B)
以上,是此公式 的证明过程,即拉乌尔根据依数性指出:对于难挥发非电解质的稀溶液,凝固点下降ΔTf都和溶液质量摩尔浓度成正比的数学证明过程。
套用领域
凝固点下降对植物的耐受性有重要意义,当外界气温发生变化时,植物体内细胞中具有多种胺基酸和强烈地生成可溶物性糖,正是这些可溶物的存在,从而使细胞的蒸气压下降,凝固点降低,保证了在一定低温条件下细胞液不致结冰;另外,细胞液浓度增大,有利于其蒸气压的降低,从而使细胞中水分的蒸发量减少,蒸发过程变慢,因此在较高的气温下能保持一定的水分而不枯萎,从而使植物表现出一定的抗旱性和耐寒性。
凝固点下降有时也称为冰点降低,套用冰点降低法还可以测定物质,尤其是高分子物质的分子量,其计算公式为
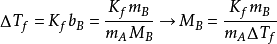
比起沸点升高法,实际套用中常使用冰点降低法进行测定,因为溶液的凝固点可以精确测定,原因有三:一是部分高分子生物样品和有机物在高温下容易破坏;二是高温下因为挥发而引起浓度变化时不能重複测定Tb;三是溶剂的凝固点降低常数比沸点升高常数大,测定结果的精确度较高。
有机化学还用测定沸点的升高和凝固点的下降来检验化合物的纯度,这是因为含杂质的化合物可以看作是一种溶液。
渗透压
现象内容
产生渗透作用有两个必要条件:一是有半透膜存在;半透膜两侧存在浓度差。在如右图所示的容器中,左边盛纯水,右边盛糖水,连通容器中间安装一种小(溶剂)分子可通过,大(溶质)分子却不能通过的具有选择性的半透膜(semi-permeable membrance),如羊皮纸、火棉胶膜、玻璃纸、动植物细胞膜、毛细血管壁等物质都具有半透膜的性质。
开始时,a容器两侧液面等高。经过一段时间以后,可以观察到b容器左侧纯水液面下降,右端糖水液面升高,说明存水中有一部分水分子通过半透膜进入了溶液,这种溶剂分子透过半透膜进入溶液或者从稀溶液进入浓溶液的自发过程称为渗透。在一定温度下,如果在溶液液面上施加压力如c容器所示,是两边液面重新持平,这时水分子从两边穿过的数目完全相等,在此达到渗透平衡。如果改用同种物质的两种不同浓度的溶液,较浓溶液的一面也不断上升,这说明水分子透过半透膜进入溶液或者从稀溶液进入浓溶液的一面。
如果半透膜两侧溶液的浓度相等,则渗透压相等,这种溶液称为等渗溶液。如果半透膜两侧的浓度不相等,其渗透压不等,则渗透压高的称为高渗溶液,渗透压低的称为低渗溶液。
在医学上把溶液中产生渗透作用的各种溶质粒子,称为渗透活性物质。由于生物体内各部位温度变化幅度不大,故医学上常用渗透浓度来衡量溶液渗透压的大小。渗透浓度就是渗透活性物质的物质的量除以溶液的体积,用符号COS表示,其常用单位也是mol/L和mmol/L,医学上的单位是Osmol/L(渗量/升)或mOsmol/L(毫渗量/升)。1 mmol/L≈1 mOsmol/L。在计算溶液的渗透浓度时应注意,对于强电解质溶液,其渗透浓度等于溶液中溶质的离子总浓度;对于弱电解质,其渗透浓度等于溶液中未解离的非电解质分子的浓度和解离出的离子浓度的总和;而对于非电解质,其渗透浓度等于其溶液浓度。
现象解释
渗透现象产生的原因可解释为:由于膜的两侧水的摩尔分数不等,溶液蒸发逸出的水分子较少,即溶液的蒸气压小于纯溶液的蒸气压。所以纯水分子通过半透膜进入溶液的速率大于溶液中分子通过半透膜进入纯水的速率,即单位时间内从纯水进入溶液的水分子数要比从溶液进入纯水的多。
然而随着渗透的进行,溶液端水柱逐渐升高,水柱产生的静水压使得单位时间内进、出的水分子数目渐趋接近,一旦相等时,体系建立渗透平衡,此时为了阻止渗透作用进行,阻止溶剂进入溶液而必须向溶液施加的最小压力称为渗透压,用符号П表示。这里的渗透压是用膜两面的液面高度差所产生的压力(F=ρgh)来量度,其数值等于渗透达到平衡时液面高度所产生的静水压。
有学者指出,渗透压为维持只允许溶剂通过的膜所隔开的溶液与纯溶剂之间的渗透平衡而需要的超额压力说法不能作为定义,即无所谓施加这一说,这只是只是测定渗透压大小的方法。溶液的渗透压的定义应该是连通器两边平衡状态下的压差。因为渗透压是溶液本身固有的性质之一,是通过膜指向溶液的单位面积上的力,不管是否额外施加,也不管半透膜间隔的对象溶液的另一边溶液的浓度是多少,无论有无半透膜,它都始终存在,只是不一定表现出来,且为定值。
渗透压定律
1866年,荷兰物理学家范特霍夫(van't Hoff)总结大量实验指出,在一定温度下,难挥发非电解质溶液的渗透压与溶质B的物质的量成正比,当浓度不变时,稀溶液的渗透压P和热力学温度T成正比,比例係数为摩尔气体常数,写成数学式为:
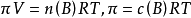
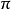
当溶液很稀时,则有: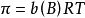
稀溶液的渗透压和其他依数性还可以在数值上联繫起来,即
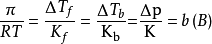
数学推导
此时溶液中溶剂组分的化学势与纯溶剂一侧的化学势相等,渗透压达到平衡。
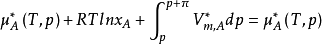
假定纯溶剂的摩尔体积不随压力变化,积分上式:
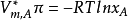
对于各组成物质在全部浓度範围内都服从拉乌尔定律的溶液,称之为理想溶液,此式是理想溶液的渗透压公式。
对稀溶液有: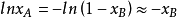 ,式子变为:
,式子变为:
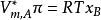
若取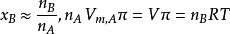 ,则有
,则有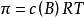
此式被称为渗透压公式,渗透压的大小与溶质的量有关,而与溶质的性质无关。
套用领域
溶液的渗透压在生物学中有很重要的作用。鲜花插在水中,可以数日不萎缩;海水中的鱼不能在淡水中生活,淡水鱼不能在海水中养殖,都与渗透压有关。
植物的细胞壁有一层原生质,起着半透膜的作用,而细胞液是一种溶液,其渗透压可高达2.0×10 Pa,土壤中水分通过这种渗透作用,送到树梢。当植物处于水分充足的环境中,水通过半透膜想细胞内渗透,是细胞内产生很大的压力,细胞发生膨胀,植物的根茎叶和花瓣就会有一定的弹性,这样植物就能更好地向空间伸展职业,充分吸收二氧化碳和接受阳光。如果土壤溶液的渗透压高于植物细胞液的渗透压,就会造成植物细胞液内的水分向外渗透,导致植物枯萎。农业生产上改造盐硷地、合理施肥和施肥后及时灌水就是这个道理。

另外,人体组织内部的细胞膜、血球膜和毛细管壁都具有半透膜的性质,而人体的体液,如血液、细胞液和组织液等都具有一定的渗透压,对病员人体静脉输液时,必须使用与人体血液的渗透压相等的溶液,在310 K时,渗透压约为7.7~7.8×10 Pa,如临床常用的0.9%胜利盐水和5%的葡萄糖溶液。否则有渗透作用,可以引起血球膨胀或萎缩而产生严重后果,右图为红细胞在不同浓度的NaCl溶液中的形态示意图。当因发烧或其他原因,人体内水分减少时,血液渗透压增高,即产生无尿、虚脱等现象,故应多饮水以降低血液的渗透压。
毛细血管壁也是一种半透膜,隔着血浆和组织间液,它能让低分子的水、葡萄糖、尿素、胺基酸和离子自由通过,因此,血液和组织间液间渗透压差及水盐平衡取决于胶体渗透压,如果因某种原因导致血浆蛋白质减少,血浆的渗透压降低,血浆中的水分子和其他小分子、离子就会透过毛细血管壁进入组织间液,导致血容量(人体血液总量)降低,组织间液增多,这是形成水肿的原因之一。临床上对大面积烧伤或由于失血过多而造成血容量降低的患者进行补液时,除补以生理盐水外,还须同时输入血浆或右旋糖酐等代血浆,才能恢复血浆胶体渗透压和增加血容量。
工业上常常利用渗透的对立面-反渗透来为人类服务。所谓反渗透,就是在溶液上加一个额外的压力,如果这个压力超过了溶液的渗透压,那幺溶液中的溶剂分子就会透过半透膜向纯溶剂一方渗透,使溶剂体积增加,这一过程叫做反渗透。反渗透原理在工业废水处理、海水淡化、浓缩溶液等方面都有广泛套用。儘管目前成本是城市自来水生产的三倍左右,但要比用蒸馏法从海水製取淡水的能量少很多,仅为蒸馏法的30%,已成为一些海岛、远洋客轮、某些缺少饮用淡水的国家获得淡水的方法。反渗透法处理无机废水,去除率可达90%以上,有的竟高达99%。对于含有机物的废水,有机物的去除率也在80%以上。作为反渗透的物质有醋酸纤维素膜、尼龙66、聚碸醯胺膜,以及氢氧化铁、硅藻土製成的新型超过滤膜等等。
渗透压法测定非电解质的相对分子质量尤其独特的优点,其计算公式:
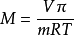
挥发性溶质溶液的依数性
要使稀溶液遵循依数性规律,溶质一定是非挥发的。但若溶质是挥发性的,又可分为下列几种情况:
溶质挥发性大于溶剂挥发性
若溶质B是非挥发性的,此时的情况如图1中曲些Ⅰ示。假若加入的溶质B的挥发性大于溶剂的挥发性,则溶液的蒸气压就应该是溶剂的蒸气压加上溶质的蒸气压,即p=p*+p(B),则溶液的蒸气压高于纯溶剂的蒸气压,使得溶液的蒸气压在较低的温度下达到latm。而使沸点下降为右图中Ⅱ所示,这是因为蒸气压上升违反了依数性规律。例如在水中加入少量乙醇,乙醇是易挥发物质,其蒸气压比同温度下的水的蒸气压大。因此加入乙醇,溶液的蒸气压增大,沸点降低,凝固点升高。
溶质挥发性小于溶剂挥发性
溶质B相对于纯溶剂A挥发性较小,溶质B也可以看作是A的溶剂。当溶液为理想或对理想溶液偏差不大时,即不论溶液的浓度如何,总能符合拉乌尔定律和亨利定律,此时对溶质和溶剂的区分已无意义。对于理想液态混合物,对任一组分的蒸气压为:p=p*x。
pA=pA*xA,pB=pB*xB,由于溶质B的挥发性较小,pB*<pA*
pB*xB<pA*xB=pA*(1-xA)=pA*-pA*xA
pA*>pA*xA+pB*xB=pA+pB=p
其中pA、 pB分别为A 与B的分压 ,pA*和pB*分别为A和B的饱和蒸气压,xA和xB为A与B的摩尔分数,p为溶液的蒸气压,pA*>p表明溶液的蒸气压必定低于纯溶剂的蒸气压,还是符合乌拉尔定律的前提,其依数性还是蒸气压不断下降和沸点不断上升。
但在这种情况下沸点上升与溶液中溶质的质量满足关係式 其中xB1、xB分别为气相、液相的溶质的摩尔分数,而不是原先的线性关係。如在苯液中加入甲苯,由于甲苯相对于苯来讲挥发性要小所以随着甲苯的加入,溶液的蒸气压下降,沸点上升,凝固点下降,这种关係可由苯-甲苯相图可看出。
其中xB1、xB分别为气相、液相的溶质的摩尔分数,而不是原先的线性关係。如在苯液中加入甲苯,由于甲苯相对于苯来讲挥发性要小所以随着甲苯的加入,溶液的蒸气压下降,沸点上升,凝固点下降,这种关係可由苯-甲苯相图可看出。
除此之外,沸点和凝固点的变化还可能随着加入的溶质的量而变化,这是由于当溶质对理想溶液发生偏差所导致的。
正偏差
当溶质对理想溶液发生正偏差时,异分子间的排斥倾向起了主导作用,因而使分子更容易从溶液中逸出。相图形状如右图所示,在相图中有最低点,如图中c点,这就决定了该类溶液的沸点变化的特殊情况,从图中可看出A的沸点比B的沸点高,即B物质的挥发性较强。
从纯净的挥发性A物质到在C点所对应的组成的区间内,当加入相对于溶剂挥发性较强的溶质B后,溶液组分中的排斥作用不断增强,溶液的沸点是下降的;而在C点所对应的组成到纯净的挥发性B物质的区间内,随着溶质B的大量地加入,溶液反而显得更加纯净,排斥作用在减弱,此时沸点是上升的。
很明显,当加入相对于溶剂挥发性大的溶质后溶液的沸点变化恰与上述讨论相反。如丙酮一CS2体系,C2H5OH一C6H6(苯)体系等的相图即可说明。此处讨论已将稀溶液扩展为溶液的範畴。
负偏差
当溶质对理想溶液发生负偏差时,组分分子间互相作用或缔合,因而使分子从溶液中逸出的能力降低。体系的相图如右图所示。相图中出现了最高恆沸点D点,因此D点决定了溶液沸点的变化。从图中可看出A的沸点比B的沸点低,即A物质的挥发性较强。
从纯净的A物质到在D点对应的组成的区间内,当加入相对于溶剂挥发性较弱的溶质B后,由于组分之间的相互作用或缔合作用不断增强,溶液更加稳定,溶液的沸点升高;在D点所对应的组成到纯净的B物质区间内,加入相对于溶剂挥发性较小的物质后,沸点上升很明显。
当加入相对于溶剂挥发性大的溶质后溶液的沸点变化恰与上述讨论相反。如硝酸—水体系,丙酮—CHCl3 体系等的相图即可说明。
电解质溶液的依数性
难挥发性非电解质稀溶液的四个依数性都能很好地符合拉乌尔定律,其实验测定之和计算值基本相符。但电解质溶液的依数性却极大地偏离了拉乌尔定律。电解质稀溶液依数性偏大的原因,是电解质在水溶液中能够完全解离,是同浓度的电解质比非电解质含有更多的溶质粒子数,这种现象称为电解质的“反常行为”,见下表。
电解质 | b(B)/(mol·kg-1) | △Tf(计算值)/K | △Tf'(实验值)/K | i=实验值/计算值 |
NaCl | 0.5 | 0.929 | 1.692 | 1.82 |
0.1 | 0.186 | 0.346 | 1.86 | |
0.05 | 0.0929 | 0.176 | 1.89 | |
0.01 | 0.0186 | 0.0361 | 1.94 | |
KNO3 | 0.5 | 0.929 | 1.414 | 1.52 |
0.2 | 0.372 | 0.664 | 1.78 | |
0.1 | 0.186 | 0.333 | 1.79 | |
0.05 | 0.0929 | 0.172 | 1.85 | |
0.01 | 0.0186 | 0.0359 | 1.93 | |
MgSO4 | 0.5 | 0.929 | 1.018 | 1.10 |
0.1 | 0.186 | 0.242 | 1.30 | |
0.05 | 0.0929 | 0.129 | 1.39 | |
0.01 | 0.0186 | 0.0300 | 1.61 | |
K2SO4 | 0.1 | 0.186 | 0.454 | 2.44 |
0.01 | 0.0186 | 0.0521 | 2.80 | |
MgCl2 | 0.1 | 0.186 | 0.519 | 2.78 |
参考资料来源
实验表明,电解质稀溶液蒸气压下降值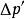 、沸点上升值
、沸点上升值 、凝固点下降值
、凝固点下降值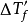 和渗透压
和渗透压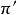 ,并且存在下列关係:
,并且存在下列关係:
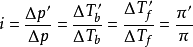
虽然强电解质在水中全部离解,强电解质的依数性理论上应是同浓度非电解质稀溶液的整数倍,但实验测得电解质稀溶液的范特霍夫校正係数i并非整数。1:1型电解质如NaCl的稀溶液的i值接近于2,1:2型或2:1性电解质如MgCl2和Na2SO4等稀溶液的i则大于2而接近于3,其余类推。且溶液越稀,i值越接近于整数。这是由于电解质在稀溶液中发生离解,而稀溶液的依数性只取决于溶质B的粒子数,而与溶质的类型无关。第一个合理解释i值并非整数的是阿侖尼乌斯(Arrhenius)的电离理论。
电离学说
1884年瑞典化学家阿侖尼乌斯(Arrhenius)根据以上实验事实,提出了电解质溶液的电离学说,用于解释电解质溶液对拉乌尔定律的偏离行为。他认为电解质溶于水后可以自发解离成阴、阳两种带电粒子,即离子,而使溶液中溶质的粒子总数增加,导致了校正係数i总是大于1;由于正负离子不停地运动,相互碰撞时又结合为分子,所以在溶液电解质只是部分电离,电离百分数称为电离度(degree of ionization)。
阿侖尼乌斯认为,若溶质是电解质,则其质点数将因电离而增加,所以△Tf'等依数性数值就会增大。例如0.10mol·kg-1的NaCl部分离解成Na+和Cl-,从而导致溶液中质点数增加。设电离度为α,则1kg溶剂中含有0.10(1-α)分子、αmolNa+、αmolCl-1,总共有0.10(1+α)mol的质点。
电离方程式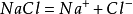
平衡时浓度/(mol·kg-1) 0.10(1-α) 0.10α 0.10α
因凝固点下降ΔTf都和溶液质量摩尔浓度成正比,有下列比例关係,右边是0.10mol·kg的NaCl△Tf'/℃的计算值和实验值。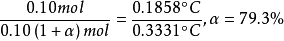
根据电离学说,计算得到0.110mol·kg-1的NaCl只有79.3%电离成Na+和Cl-,人们将这个电离度称为表观电离度(apparent ionization degree),阿侖尼乌斯认为是不完全电离导致了校正係数并非是个整数。
阿侖尼乌斯还用电导法、依数性等不同的方法测定了各种电解质溶液的电离度,如下表所示,发现依数性和导电性测定值的差别已超出了实验允许无法範围,且浓度越大差别越大。这是因为电导法和依数法都有缺陷。电导固然与溶液里离子的多少有关,但离子的电荷、离子间的相互作用、离子迁移的速率对电导都有影响,而各种离子迁移速率都是不相同的。而依数性是将带电子的离子和不带电的中性分子同等看待,后来人们发现并不存在这种不带电中性分子。
活度与离子强度
活度
虽然实验测的强电解质的电离度小于100%,路易斯(F.M.Lewis)认为,这并不意味着强电解质中一定有未电离的分子存在。非理想溶液之所以不符合拉乌尔定律,是因为溶剂与溶质之间有相当複杂的作用,在没有弄清这些相当複杂的作用之前,可根据实验数据对实际浓度(x,b,c等)加以校正,并于1907年提出了有效浓度,即活度的概念,用符号α表示:
式中:γ为校正係数,称为活度係数。因为浓度比了标準态浓度cΘ、bΘ,cΘ=1 mol·L-1,bΘ=1 mol·L-1,所以α和γ是量纲为1的数。当溶液无限稀时,离子间相互作用,可以忽略不及。此时可近似认为γ=1,即α=c/cΘ。
应当指出,用活度表示有效浓度,对非电解质的溶液,甚至对某些体系中的溶剂也可适用,活度係数γ是一个校正係数,在电解质溶液中一般是小于1的常数,但在某些特殊情况下可能大于1;在非电解质溶液中,γ可能大于1,也可能小于1。
浓度/(mol·kg-1) | HCl | KCl | NaCl | NaOH | H2SO4 | CaCl2 | CdSO4 |
0.005 | 0.928 | 0.927 | 0.929 | ---- | 0.639 | 0.785 | 0.50 |
0.01 | 0.904 | 0.901 | 0.904 | 0.89 | 0.544 | 0.725 | 0.40 |
0.05 | 0.830 | 0.815 | 0.823 | 0.82 | 0.340 | 0.57 | 0.21 |
0.10 | 0.796 | 0.769 | 0.778 | 0.776 | 0.265 | 0.524 | 0.17 |
0.20 | 0.767 | 0.718 | 0.735 | 0.757 | 0.209 | 0.48 | 0.137 |
0.50 | 0.757 | 0.649 | 0.681 | 0.735 | 0.154 | 0.52 | 0.067 |
1.00 | 0.809 | 0.604 | 0.657 | 0.757 | 0.130 | 0.71 | 0.041 |
2.00 | 1.011 | 0.576 | 0.670 | 0.70 | 0.124 | 1.55 | 0.035 |
3.00 | 1.32 | 0.571 | 0.710 | 0.77 | 0.141 | 3.38 | 0.036 |
4.00 | 1.76 | 0.597 | 0.791 | 0.89 | 0.171 | --- | --- |
参考资料来源
然而,在电解质溶液中,正负粒子总是同时共存,实际上无法用实验方法测定单种离子的活度,为此有引入了平均活度的概念。其定义式为 :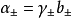
对于化学式为AxBy的强电解质溶液来说,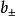 与b的关係为:
与b的关係为: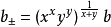
式中, 离子平均质量摩尔浓度。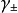 称为离子平均活度係数。它可用多种实验方法测定。它的大小不仅与离子的浓度有关,还和离子的电荷相关。实验结果表明,离子电荷对活度的影响比离子浓度还要大些。
称为离子平均活度係数。它可用多种实验方法测定。它的大小不仅与离子的浓度有关,还和离子的电荷相关。实验结果表明,离子电荷对活度的影响比离子浓度还要大些。
离子强度
路易斯于1921年提出离子强度的概念,表示离子电荷和离子浓度这两个因素对活度係数的影响:
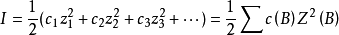
式中:I为离子强度,c(B)和Z(B)分别为组分B的浓度(包括x,b,c等)和电荷数。显然离子浓度越大,离子电荷越高,离子强度越大。
如计算0.050mol·L-1AlCl3溶液中的离子强度。
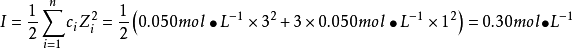
强电解质理论
1912年X射线结构分析确定电解质NaCl晶体有NaCl由Na+和Cl-组成,不存在NaCl分子。1923年,德国化学家德拜(Debye)和休克尔(Hückel)针对强电解质溶液依数性发生偏差的事实以离子键存在着相互牵製作用为基础,提出强电解质溶液理论——离子互吸理论。
强电解质在水中是完全离解的,在溶液中的粒子浓度很大。由于离子间存在较强的静电引力,对某一正离子来说,必然吸引负离子而排斥正离子,使其周围聚集较多的负离子和较少的正离子,即在正离子周围形成一个负离子的包围圈,称为离子氛(ion atomosphere),由于离子氛的存在,溶液中的粒子相互牵制,离子的运动不能完全自由,是粒子在溶液中迁移速度减慢。相当于离子减少,电离度降低。
此外,人们还发现,在强电解质溶液中不但有离子氛存在,而且带相反电荷的离子键还能相互缔合成“离子对”,1926年提出的卜耶隆离子缔合理论,认为在溶液中凡是异号电荷离子间静电吸引能超过2kT,静电能大于热运动能时,即可形成离子对。如Na+Cl-,在溶液中作为一个整体运动,比较稳定,就像一个电中性分子。
由于“离子氛”和“离子对”的影响,强电解质溶液的依数性都比完全离解的理论计算值小,溶液越浓或离子价数越高,这种偏差就越大。
德拜-休克尔极限公式
离子平均活度係数 与离子强度I有关
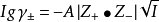
如计算25°C时,计算0.01mol·kg-1的NaCl溶液中,离子的平均活度係数和离子的平均活度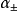 。
。
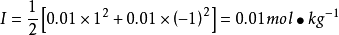
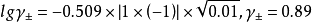
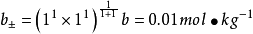
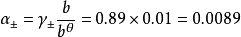
德拜-休克尔公式
下式为德拜-休克尔公式,可以计算单个离子的的活度係数,适用在稀溶液(c<0.Imol·L-1):
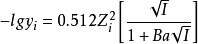
其中B、a为常数,Zi为离子的电荷;I为溶液的离子强度。
如计算0.050mol·LAlCl3溶液中Cl-1和Al3+的活度係数并由此计算活度。已知Cl-1的a=300,Al的a=900,b=0.00328,I=0.30mol·L-1。由德拜-休克尔公式:
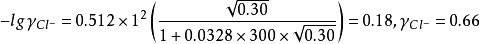
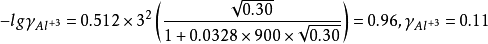
由此可以看出同样的离子强度,对高价离子影响大,其离子活度a会极大地下降。
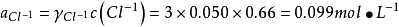
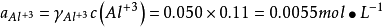
套用领域
电解质溶液依数性的套用也有很多。例如,工业上或实验室中常採用某些一潮解的固态物质如CaCl2、P2O5等作为乾燥剂,其乾燥原理是蒸气压下降:固态物质潮解后在其表面形成饱和溶液,由于溶液浓度较大,其蒸气压较低,当空气中水蒸气的分压大于溶液的蒸气压时,空气压重的水蒸气就会不断地进入溶液,使固态物质不断潮解,直到固体物质完全溶解或气液两相的蒸气压相等为止。
利用溶液凝固点下降这一性质,冬季可撒盐除雪,当食盐或氯化钙与冰(或雪)接触时,在食盐或氯化钙的表面形成极浓的盐溶液,而这些浓盐溶液的蒸气压比冰(或雪)的蒸气压低得多,冰(或雪)则以升华或熔化的形式进入盐溶液。进行上述过程都要吸收大量的热,从而使体系的温度降低。利用这一原理,可以自制冷冻剂。一份食盐和三块冰混合,体系的温度可降至-20℃,10份六水氯化钙与7~8份碎冰均匀混合,体系的温度降至-40~-20℃。用CaCl2、冰和丙酮的混合物,可以致冷到-70C°以下。
| 盐 | 质量/g | 冰盐点/℃ |
|---|---|---|
KOH | 47.1 | -65.0 |
ZnCl2 | 108.3 | -62.0 |
CaCl2·6H2O | 143.0(125.0) | -55.0(-40.3) |
NaCl | 33 | -21.2 |
NaNO3 | 59 | -18.5 |
NH4Cl | 25 | -15.8 |
KCl | 30 | -11.0 |
CaCl2 | 80 | -11.0 |
参考资料来源
浓溶液的依数性
浓溶液由于存在着更为複杂的情况,导致其依数性与理想情况偏离较为严重,所以依数性跟计算值有差异。但依然认为其具有依数性的。高浓度使得溶质粒子之间的相互影响大为增加,简单的依数性的定量关係不再适用。
溶剂化
这是因为溶液中溶质粒子是溶剂化的,溶剂化程度的大小与粒子种类、溶液浓度、温度等因素有关;液体的结构是最複杂的。 液体与气体一样具有流动性, 但具有体积这一点却与固体相似。由液体的流动性可知, 它与气体一样粒子间相对运动激烈, 因此粒子的排列不规则; 从它的密度接近固体这一点看, 又不象气体那样紊乱。据X-射线研究已知: 液体不是完全无规则, 而具有某种程度的与结晶性固体相似的有规则的分子排列, 其规律性离中心分子仅有几个分子程度。且已明确了其排列只限于短程式,而不象晶体那样的长程式。 可见: 溶剂分子间往往是缔合的, 而不是单个的“质点”。缔合程度的大小与溶剂的种类和温度有关。
粒子半径
拉乌尔定律未考虑溶质和溶剂粒子的大小差别。首先, 由于溶剂分子间始终存在着缔合与解离之间的动态平衡, 每个“ 溶剂粒子”( 指同缔合程度的溶剂分子的混合物)的大小就不同了。这种因缔合程度不同引起的“ 溶剂粒子”大小的差别是动态变化的、难以準确测定的。其次, 不同溶质粒子本身大小差别很大。 从鲍林推算出的一些常见离子半径以看出, 即使是单原子离子的半径差别也可高达数倍。葡萄糖、蔗糖、环糊精等多原子分子由于有数十个甚至上百个原子组成, 其半径必然是水分子等小分子半径的数倍。 高分子化合物溶液中的溶质与溶剂分子的半径差别可达数十倍甚至上百倍。 这种粒子大小的差别是不可忽略的。第三, 由于溶剂化作用的存在, 同一溶液在不同浓度和温度下溶质粒子的溶剂化程度不同, 粒子大小也就不同了。当浓度一定时, 温度升高则每个溶质粒子的溶剂化程度减小; 当温度一定时, 浓度增大则每个溶质粒子的溶剂化程度减小。 而且由于分子的热运动, 每个溶质粒子的溶剂化程度的大小是动态变化的。
分子极性与范德华力
再者,电子的被发现,共价键 、分子的极性及范德华力等理论的形成,使人们认识到:极性也是非电解质分子的本性之一,受分子极性的影响,不同分子间的范德华力也不相同,极性分子和极性分子之间以较强的取向力为主进行相互作用,有的甚至可以形成较强的氢键,非极性分子和非极性分子之间以较强的色散力为主进行相互作用,极性分子和非极性分子之间以较弱的诱导力为主进行相互作用。在那个年代,拉乌尔不可能考虑到极性也是非电解质分子的本性之一,而在讨论浓溶液时,就不能忽略溶液中溶质粒子的溶剂化作用,溶剂粒子与溶剂粒子间 、溶剂粒子与溶质粒子间 、溶质粒子与溶质粒子间的范德华力等方面的问题。
所以,此时依数性的定义中说与“溶质的本性无关”就不对了。